【本報訊】患有先天性巨結腸症的嬰兒大腸不會蠕動,兩成人因病情嚴重至要切除大部份腸臟,甚至因大便淤塞,爆腸奪命。香港大學的研究為治療帶來新希望,研究發現患者先天有兩組基因突變,影響腸道神經幹細胞發展,失去控制正常排便功能;專家預料將來可用針對該兩組基因的藥物,配合幹細胞移植,代替現有的切腸治療。 記者:陳凱迎
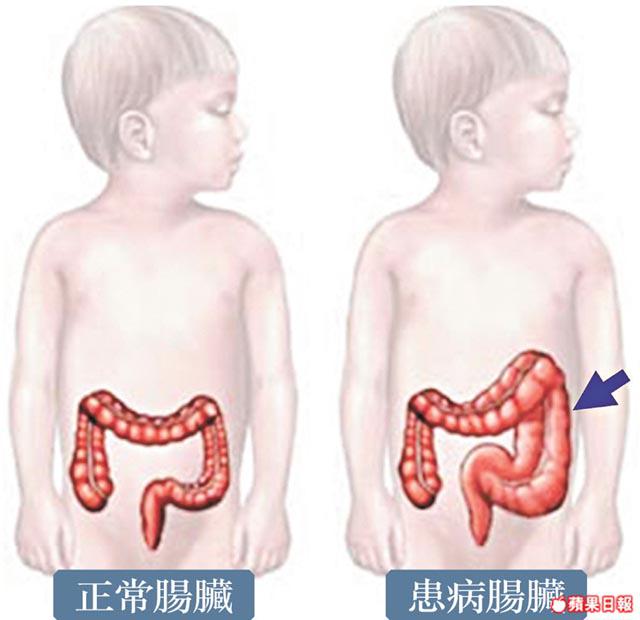
在亞洲,每3,000名初生嬰兒便有一人患有先天性巨結腸症(Congenitalmegacolon),患者腸道因先天缺乏神經細胞,不會蠕動,患此症的初生嬰兒會有嚴重便秘。港大外科學系臨床助理教授黃格元指,患者一歲前已發病,可利用X光造影檢查及活組織化驗確診,八成患者受影響的腸道範圍較短,病情較輕,但餘下病人會因大便淤塞,令腹部腫脹,甚至有爆腸危機。
一人病逝一人留醫
本港過去10年,逾120名患有此病的嬰兒需進行手術,將有問題的腸道透過微創手術切除;但上述嚴重患者中,有部份人要將整條腸道切除,往後因無法正常吸收,須長期住院,靠營養液續命;以瑪麗醫院為例,過去半年便有兩名嬰兒,因全腸缺乏神經細胞,須切除大部份腸臟,其中一人病逝,另一人留醫。
缺幹細胞影響排便
醫學界近年先後發現多個與先天性巨結腸症相關的基因,港大外科學系助理教授顏秀慧指,正常的排便功能,包括腸道蠕動,須透過腸道的神經元細胞和膠質神經細胞相互配合;最新的研究在小鼠及人類皮膚細胞內發現,先天性巨結腸症患者體內,因兩組分別名為PTCH1及DLL3的基因突變,令腸道神經幹細胞提早發展成膠質神經細胞,結果沒有足夠的幹細胞發展成神經元細胞,破壞整個排便功能。
顏說,新的研究發現有助改善現時僅有的切腸治療。她預計,8至10年後醫學界可利用幹細胞移植技術,替先天性巨結腸症患者補充不足的幹細胞,配合針對上述兩組基因的藥物,令移植的幹細胞變成神經元細胞,以恢復正常的排便功能。有關研究結果已於學術期刊《JournalofClinicalInvestigation》發表。